The EU Medical Device Regulation 2017/745 (MDR) entered into force on 26 May 2017 and began to apply from 26 May 2021, superseding the EU Medical Device Directive 93/42/EEC (MDD) and the Active Implantable Medical Device Directive 90/385/EEC (AIMDD). Although there are many similarities between the two legislation, the MDR presents a key update for the regulation of medical devices in the EU, laying down new requirements for all stakeholders involved.
Some key updates include:
- Greater focus on Post-Market Surveillance.
- Eudamed.
- Unique Device Identifiers.
- New classification rules.
- Person Responsible for Regulatory Compliance.
- The General Safety and Performance Requirements.
Overall, there is increased scrutiny on the industry’s ability to meet the new regulations, with specific focus on technical documentation and clinical evidence of safety and performance. The new Regulation aims to improve the high standards of quality and safety of medical devices to ensure that patients are receiving.
Due to the COVID 19 pandemic, the original date of application was postponed by one year to 26 May 2021. Further to this, an amendment to Article 120 allowed for certain devices to qualify for an extended transition period. To read further on this subject, check out our Article 120 information page.
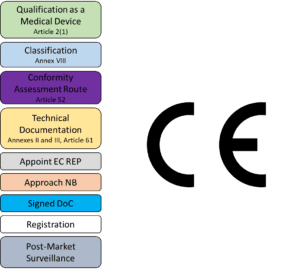
The first step in entering in placing your product on the EU market as a medical device is to verify that this product qualifies as a medical device. This should be done by directly consulting the definition of a medical device in Article 2(1) of the MDR or by referring to MDCG 2024 Guidance on classification of medical devices. Either way, manufacturers should record their rationale as to why this product is considered a medical device.
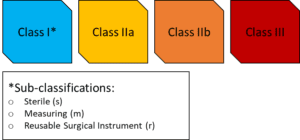
The extent of the application of the requirements within the MDR will greatly depend on the Classification of the medical device. Devices are classified in 4 distinct risk categories, in increasing order of risk: Class I, Class IIa, Class IIb, and Class III. To determine the risk classification of your device, you would be expected to look into Annex VIII of the EU MDR which lays down ‘Rules’ which you can use to find the appropriate match. We suggest that you also consult MDCG 2021-24 which may be helpful in this regard.
Following the correct classification of your product, you will need to find out what Conformity Assessment Procedure will be applicable to your device, as found in Article 52. Depending on your classification and your choice, you will be able to determine which other sections of the MDR will be applicable to your device.
The EU Member States designate third-party, external organisations called Notified Bodies (NB) to conduct conformity assessments of medical devices. A NB is not required by all devices, but is required for Class IIa, IIb, III devices as well as Class I devices which are sterilised, have a measuring function, or are reusable surgical instruments. Therefore, for “simple” Class I devices, there will be no NB involvement; these are often called self-declared devices. For such Class I devices, the requirements of the MDR will still apply, however, NB will not be involved in scrutinising your conformity with respect to the MDR.
All medical devices will be required to comply with the technical documentation requirements as laid down in Annexes II and III of the EU MDR. The detail required in these sections will largely depend on the classification, overall device risk, and novelty of the device itself; however, it is important to understand that all devices must have this technical documentation. Read more about Technical Documentation, here, and see how our consultants can help you with putting these together.
Within the context of the MDR, an Economic Operator is either a legal manufacturer, authorised representative, importer, or distributor. Depending on which of these your organisation falls under, you will need to meet the obligations laid out for each of these within the MDR.
If you are a legal manufacturer based outside of the EU, you will be required to appoint an EU Authorised Representative which has obligations in verifying that you are meeting your regulatory requirements and will be your contact point in the EU. Advena Ltd. has been providing this service for over 25 years. Read more about this service, here.
Furthermore, manufacturers must identify an importer based in the EU for the devices being placed on the EU market.
The obligations of manufacturers are laid out in Article 10 of the MDR. One of these obligations is that each manufacturer must operate under a Quality Management System (QMS); this is to ensure that the devices being placed on the market are safe and compliant. An audit of your QMS will be required for higher-risk classifications, however, simple Class I device may operate a QMS without the need to certify against a particular standard. For more information on QMS, see our information page specifically on this topic.
Unique Device Identifier (UDI)

From 26 May 2021, all devices compliant with the EU MDR will need to be assigned a Unique Device Identifier (UDI). The UDI system is explained in further detail on our UDI page. Although there is a stepped transition period for the provision of the UDI on the device labelling, from 26 May 2021, all MDR-compliant devices will need to be assigned a UDI-DI and a Basic UDI-DI. The Basic UDI-DI is the ‘primary identifier’ for a given product family.

The UDI-DI and Basic UDI-DI may be generated in accordance with the standards of designated UDI-issuing entities and shall be displayed on the Declaration of Conformity.
Registration

Manufacturers, Authorised Representatives, and Importers will also be required to register their organisations on Eudamed in order to obtain their Single Registration Number (SRN). Furthermore, manufacturers will be obliged to register all their medical devices on Eudamed; this involves the registration of their UDI-DI and Basic UDI-DI for all product variants. Eudamed will replace the previous system whereby registrations were performed through direct communication with the National Competent Authority.
